|
Size: 2853
Comment:
|
Size: 3486
Comment:
|
| Deletions are marked like this. | Additions are marked like this. |
| Line 61: | Line 61: |
| Line 62: | Line 63: |
| Coming soon. | * '''Do I really need a GPU?''' Technically, no. In practice, yes. On a modern GPU, the code runs in an hour or less. On the CPU, it depends on the number of threads, but it can easily take a whole day. * '''What are the two additional arguments listed on the help?''' You should not need to touch these, but the 5th argument (BF_MODE) changes the set of basis functions for bias field correction and you could potentially try tinkering with it if the bias field correction fails (i.e., if bf_corrected.mgz has noticeable bias). The 6th argument is GMM_MODE, which allows you to change the grouping of ROIs into tissue classes. |
Bayesian Segmentation with Histological Atlas
This functionality is available on development versions newer than October 26th 2023
Author: Juan Eugenio Iglesias
E-mail: jiglesiasgonzalez [at] mgh.harvard.edu
Rather than directly contacting the author, please post your questions on this module to the FreeSurfer mailing list at freesurfer [at] nmr.mgh.harvard.edu
The manuscript describing this module is still in preparation:
Contents
- General Description
- Installation
- Usage
- Frequently asked questions (FAQ)
1. General Description
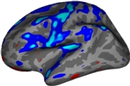
This module uses our new probabilistic atlas of the human brain to segment 333 distinct ROIs per hemisphere on in vivo scans. Segmentation relies on a Bayesian algorithm and is thus robust against changes in MRIi pulse sequence (e.g., T1-weighted, T2-weighted, FLAIR, etc). Sample slices of the atlas and the segmentation of the sample subject "bert" are shown below:

2. Installation
The first time you run this module, it will prompt you to download the atlas. Follow the instructions on the screen to obtain the files.
3. Usage
To segment a brain MRI scan,
mri_histo_atlas_segment INPUT_SCAN OUTPUT_DIRECTORY GPU THREADS
where:
INPUT_SCAN: scan to process, in mgz or nii(.gz) format.
OUTPUT_DIRECTORY: directory where segmentations, volume files, etc, will be created (more on this below).
GPU: set to 1 to use the GPU (we highly recommend using a GPU to run this module; without a GPU, running this module on a single scan can take a whole day). The GPU requirements depend on the image but are about 24GB of memory.
THREADS: number of CPU threads used by the code (set to -1 to use all available threads).
In the output directory, you will find:
bf_corrected.mgz: a bias field corrected version of the input scan.
SynthSeg.mgz: SynthSeg segmentation of the input (which we use in preprocessing and to initialize Gaussian parameters).
MNI_registration.mgz: EasyReg registration to MNI space, use in preprocessing.
seg_[left/right].mgz: segmentation into 333 ROIs of the left and right hemisphere, respectively.
vols_[left/right].csv: CSV spreadsheet with the volumes of the different ROIs, computed from the posteriors (soft segmentations).
lookup_table.txt: FreeSurfer lookup table mapping label indices to brain anatomy. You need it when visualizing the segmentations with Freeview.
4. Frequently asked questions (FAQ)
Do I really need a GPU?
Technically, no. In practice, yes. On a modern GPU, the code runs in an hour or less. On the CPU, it depends on the number of threads, but it can easily take a whole day.
What are the two additional arguments listed on the help?
You should not need to touch these, but the 5th argument (BF_MODE) changes the set of basis functions for bias field correction and you could potentially try tinkering with it if the bias field correction fails (i.e., if bf_corrected.mgz has noticeable bias). The 6th argument is GMM_MODE, which allows you to change the grouping of ROIs into tissue classes.